Levi MacIntosh (not his real name), a 38-year-old media creator in British Columbia, Canada, lost his uncle six years ago to terminal cancer that had spread to several organ systems. “He was only in his mid-fifties and hardly had any symptoms before he was diagnosed,” MacIntosh told Genetic Literacy Project. Since the cancer was so advanced and widespread, doctors had trouble determining where in the body it had originated. “It would have been great if [the doctors] had been able to tell what was happening a lot earlier. He was in the prime of his life.”
MacIntosh’s experience is an all-too familiar one for countless friends and loved ones of those diagnosed with late stage or terminal cancer. And for those who survive their own cancer diagnoses, the treatments themselves can leave behind a series of physical and emotional scars.
Long-term effects of surgical cancer treatments can include infertility, nerve damage, chronic pain, phantom limb syndrome following amputation, and lymphedema. Long-term effects of chemotherapy can include increased risk of developing leukemia, hearing loss, kidney failure, heart problems, lung disease, and immune system dysfunction. And long-term effects of radiation can include damage to the heart and lungs, development of additional cancers, troublesome or painful skin changes, and bladder and bowel control problems. It’s not uncommon for a cancer patient to receive all three forms of treatment.
Additionally, the experience of cancer diagnosis and treatment in general is associated with long-lasting psychosocial effects, as well as increased rates of depression, anxiety, post-traumatic stress disorder, and exacerbation of pre-existing mental health conditions.
Since one in three of all North Americans will be diagnosed with at least one form of cancer in their lifetimes, the impact of a reactive cancer care system is far-reaching. The ideal form of cancer care is proactive, where preventative steps are taken well before symptoms appear and where an individual’s cancer risk is known and tracked over their lifetime.

Proactive cancer care would reduce the number of lives lost, and would also minimize the financial and physical hardships associated with cancer treatments. The evolution from reactive to proactive care is only in its infancy, but research into the genes that predispose an individual to developing certain cancers helps form an important basis for future proactive cancer care measures.
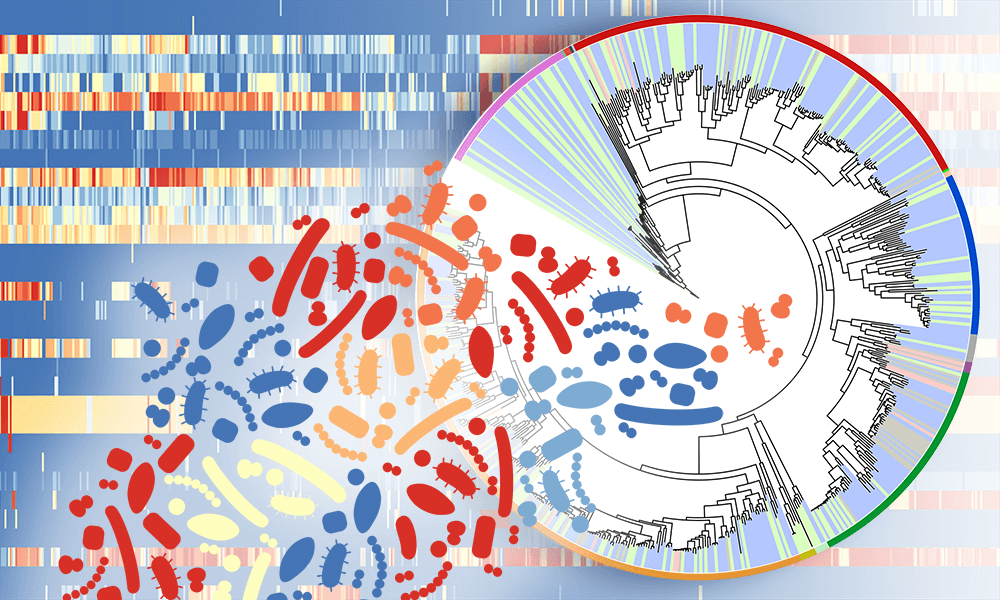
Researchers at the Centre for Genomic Regulation in Barcelona have developed a new method for identifying genes that contribute to heritable cancer risk. The new research, published in Nature Communications in July, marks a major win for big data analysis, and signifies a move in the direction of improved cancer predisposition gene discovery. It’s been estimated that five to 10 percent of all cancers are linked to inheritance of cancer predisposition genes (CPGs), but some research puts the percentage much higher — at nearly 35 percent.
“Now we have a powerful tool to detect new cancer predisposition genes and, consequently, to contribute to improving cancer diagnosis and prevention in the future,” said Solip Park, first author of the study. If a person’s cancer predisposition genes are known, screening and testing can begin long before symptoms arise, and steps could be taken to help prevent malignant tumor formation.
The team was led by ICREA Research Professor, Ben Lehner, and though they identified just 10 CPGs (and three previously-identified ones), these genes account for one in 50 of all cancer cases. ALFRED, the new statistical method that was used to discover the genes, is less expensive and more efficient than earlier methods.
The researchers relied on publicly available genome data from global cancer studies like The Cancer Genome Atlas project. ALFRED was applied to the genome sequences of over 10,000 cancer patients spanning 30 different cancer types. Genetic information from the cancer patients’ tumor cells was compared to their own non-tumor (non-cancerous) cells.
ALFRED (“allelic loss featuring rare damage”) was specifically used to test Alfred Knudson’s two-hit hypothesis, which has been a framework for CPG discovery for over four decades. The two-hit hypothesis states that for a cell to become cancerous and lead to tumor formation, both copies of the cell’s tumor suppressor genes
Genetic mutations that can contribute to the development of cancer in an individual are inherited or acquired. Acquired mutations can be caused by several factors, including DNA replication errors or UV exposure from the sun. Previous research using large-scale genome sequencing has played a big role in identifying cancers driven by the latter group, called somatic mutations, but the identification of inherited cancer risk genes has not been as productive.
For cancer to develop, according to the two-hit hypothesis, an individual needs to have a harmful inherited genetic variant (or CPG) plus the addition of an acquired somatic mutation. These “two hits” knock out the function of a tumor suppressor gene, thus blocking the cell’s abilities to keep unwanted and harmful (cancerous) cell growth in check.
The work of Lehner et al. shows that Knudson’s hypothesis can be utilized to identify CPGs from cancer exomes without the use of control groups. ALFRED, the unique statistical method used by the researchers, was able to identify CPGs that lead to cancer through a combination of inherited and somatic mutations.
What’s more, Lehner said, “our work highlights how important it is to share genomic data. … We combined data from many different projects and by applying a new computational method were able to identify important cancer genes that were not identified by the original studies. Many patient groups lobby for better sharing of genomic data because it is only by comparing data across hospitals, countries and diseases that we can obtain a deep understanding of many rare and common diseases.”
Kristen Hovet is a journalist and writer who specializes in psychology, health, science and the intersection of sociology and culture. Follow her on her website, Facebook or Twitter @kristenhovet
This article originally ran on the GLP September 25, 2018.