The latest chapter: An “open letter” in the April 7, 2021 New York Times, calling for “a full investigation into the origins of COVID-19.” The two dozen scientists who signed the letter cite the continuing absence of a “robust process” to examine critical records and biological samples. Their argument responds to the WHO’s March 20 press event that barely considered an origin other than from a natural spillover.

But two types of new information may counter the lab escapee hypothesis: filling-in-the-blanks of mammals that may have served as “missing links” in the evolution of disease transmission, and the rapid rise of viral variants reflecting a tendency to mutate that may underlie SARS-CoV-2 seemingly bursting from out of nowhere.
So here is my view, as a geneticist, of three possible origins of SARS-CoV-2:
- Bioweapon – an engineered pathogen or escape of a natural candidate
- Gradual evolutionary change through intermediate animal hosts, mutating along the way and becoming more virulent
- “Mutator” genes that trigger mutations in other genes, speeding evolution
Bioweapon beginnings
Until recently, the idea that SARS-CoV-2 was groomed as a weapon relied on a predecessor, a coronavirus dubbed RaTG13, found in the horseshoe bat Rhinolophus affinis.
Researchers discovered RaTG13 in bat droppings in an abandoned mineshaft near a cave in Yunnan, China, in 2013, shortly after six miners fell ill and three of them died of an unspecified pneumonia. (Bats harbor many viruses without becoming sick, which I explain here.)

RaTG13 shares about 96.1% of its genome sequence with that of SARS-CoV-2. For perspective, SARS-CoV-2’s genome is only about 80% similar to that of the original SARS coronavirus from 2003.
A key difference between RaTG13 and SARS-CoV-2 is in part of the receptor binding domain where the spike protein latches onto human cells. This differing part matches the RNA sequence from viruses in the Malayan pangolin, a spiny anteater-like creature that may be an intermediate host between bats and people in the infection chain. The transfer from bat to pangolin might have happened near the mine, or at a wet market rife with raw flesh, or in many other places where humans encroach on the territories of other animals and we simply haven’t looked.
Clues to the transition from bat virus RaTG13 to human virus SARS-CoV-2 may lie within the 4% of the genome sequences that diverge. Evolutionary biologists estimate it would have taken at least 50 years for the bat virus to have mutated itself into SARS-CoV-2, considering known, natural mutation rates of viral genomes. A bioweapon, presumably, could have been created much faster, just as it’s faster to buy a new car than to fix an old one part by part. But as we’ve learned, we can’t rely on what we know about past viruses. That is, the mutation rate of the newbie could be much faster than what we’ve seen before.
A document dubbed the “Yan report” (which I wrote about here) maintains that RaTG13 never existed. Instead, Li-Meng Yan and colleagues argue that the supposed SARS-CoV-2 predecessor is an imaginary RNA sequence uploaded to the GenBank database to provide a plausible natural explanation for the origin, to deflect attention from the idea of a bioweapon. The paper (there’s an initial and updated version) questions why, if RaTG13 was discovered in 2013, it wasn’t reported until February 3, 2020, in Nature. The Yan report never made it beyond preprint (non-reviewed) status, I suspect partly because of the pervasive tone of paranoia and the funding source, an organization connected to Steve Bannon. Researchers have eviscerated it, and Wikipedia reviews the details.

A brief report that I keep going back to appeared in Nature on March 17, 2020, when the global death toll from COVID stood at just 4,373: “The proximal origin of SARS-CoV-2.”
The “proximal origin” authors (Kristian G. Andersen, Andrew Rambaut, W. Ian Lipkin, Edward C. Holmes and Robert F. Garry) compare key parts of the new pathogen to corresponding parts of other coronaviruses, concluding “(o)ur analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus.” Part of their reasoning is common sense: the virus doesn’t bind to our cells strongly enough to have been invented. It’s an imperfect weapon. Why would a new iPhone work worse than its predecessors? It’s more likely, they argue, that the new virus, with its distinctions (like a dozen extra RNA bases inserted into the area corresponding to where the two parts of the spike protein attach), arose from natural selection. The virus had a natural advantage, so it was perpetuated – not invented.
Whatever happened, the prescient “proximal origin” researchers concluded, back in March 2020,
Although no animal coronavirus has been identified that is sufficiently similar to have served as the direct progenitor of SARS-CoV-2, the diversity of coronaviruses in bats and other species is massively undersampled.
That’s changing. Slowly.
Evolution in a poop soup
A leap from the RaTG13 virus found in the bat muck of the abandoned mine in 2013 to the emergence of SARS-CoV-2 in 2019 is like reading the first and last chapters of a novel: there’s not enough of a plot to reconstruct a story. But as more chapters are being revealed, it’s looking like SARS-CoV-2 arose from a poop soup of viruses – and continues to evolve.
It turns out that RaTG13 wasn’t the only stop on the evolutionary road to SARS-CoV-2. Nor was China the only home of novel coronaviruses, although they continue to be identified there. Consider recent reports:
Cambodia, January 26, 2021: Excrement and saliva from two horseshoe bats sampled in Cambodia in 2010 yielded coronaviruses that share 92.6% of their genome sequences with SARS-CoV-2, differing at one end of the gene encoding the spike protein. Concludes a preprint in bioRxiv,
The discovery of these viruses in a bat species not found in China indicates that SARS-CoV-2 related viruses have a much wider geographic distribution than previously understood, and suggests that Southeast Asia represents a key area to consider in the ongoing search for the origins of SARS-CoV-2, and in future surveillance for coronaviruses.
Thailand, February 9, 2021: Blood from five bats in a cave in Thailand yielded coronaviruses similar to a type found in Yunnan, China, as well as antibodies against SARS-CoV-2. The antibodies were also detected in a pangolin, according to a report in Nature. Although this study didn’t reveal the progenitor, it too extends the realm of SARS-CoV-2-like viruses beyond China.
China, March 8, 2021: Another bioRxiv preprint describes genome sequences of 411 coronavirus samples from 23 bat species collected from May 2019 to November 2020, over 2700 acres in Yunnan province. The closest relative to SARS-CoV-2, dubbed RpYN06, shares 94.5% of the genome sequence. But overall genome similarity is not as important as correspondence between individual genes, which can better predict the effect of a novel virus on a human body.
RpYN06 is actually the closest relative to SARS-CoV-2 identified so far, based on key genes that provide the tools to replicate (ORF1ab), melt into our cells and latch onto our protein synthetic machinery (ORF7a and ORF8), and encode the nucleocapsid (N) proteins that protect the viral genetic material. The study found 3 other coronaviruses whose genomes are very similar and resemble those found in pangolins.
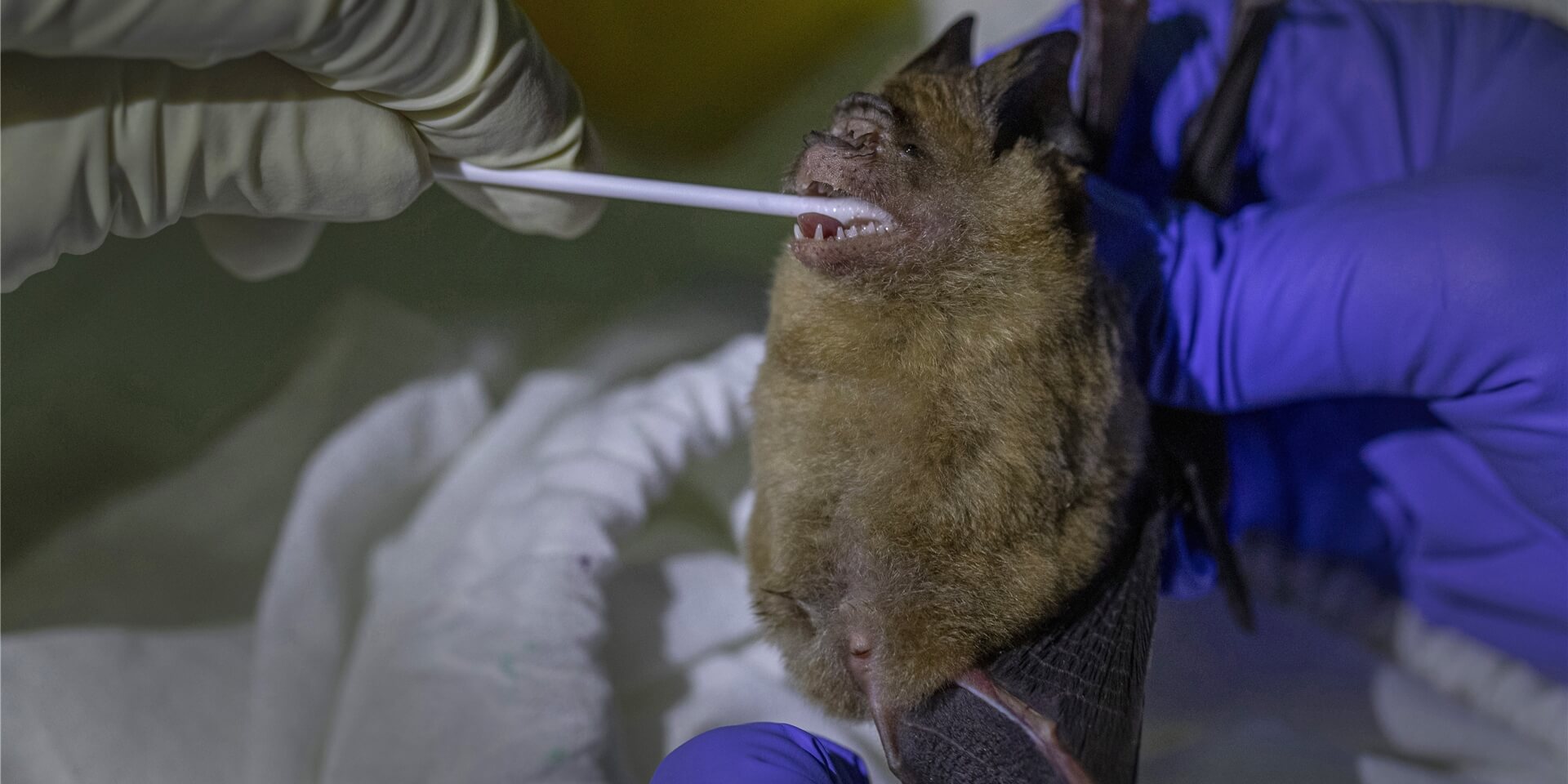
Did SARS-CoV-2 chug along happily, in various types of bats for who knows how many years, mixing with other coronaviruses and unchanging because it’s genome served it well? Only after the jump to a new host – us – did the mutations that underlie adaptation happen, spontaneously, and then persist if they conferred an advantage. Then mutations in individual genes began to accrue into the viral variants that are now sweeping the planet. The title of a recent article in PLoS Biology sums up the forces that have molded the novel coronavirus:
Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen.
The mutator hypothesis
A third way that SARS-CoV-2 could have come into being quickly is if a gene or genes has functioned as a “mutator,” provoking other genes to mutate.
I remember this phenomenon from my training as a Drosophila geneticist. Fruit flies that have a mutant yellow eye color can have offspring that revert to the normal red color, not from a mutation in an eye color gene, but from a mutation in a gene called mutator. It wreaks havoc on other genes. And the types of mutations it brings are like those in the new variants of SARS-CoV-2.

Half of the mutations that the fruit fly mutator gene causes are deletions – missing bits of genes. A telltale sign of the viral variant B.1.1.7, first detected in the UK, is “S gene target failure.” That’s when a PCR COVID test doesn’t replicate the RNA that encodes the spike protein because two amino acids are missing, while it does replicate the other viral genes.
In fruit flies, mutator also quintuples the rate of single-base changes, called point mutations. Those happen in the new viral variants too.
I’m not suggesting that a fly gene has run amok in viruses, but might a mutator-like gene be driving the rapid diversification of SARS-CoV-2 into a suite of variants? If so, rapid mutation might explain how the virus came into being and then became a shape-shifter, without having to invoke a mad-scientist-creating-a-bioweapon scenario or a string of hapless mammals passing along a pathogen that would kill millions of people in a year.
Identifying a mutator would require unraveling gene-gene interactions, something that hasn’t been a huge priority even in analyzing human genomes. Perhaps a well-studied gene of SARS-CoV-2 has a second function, inducing mutation of another? Although more than a million SARS-CoV-2 genome sequences have been uploaded to the database GISAID, I don’t know to what extent researchers are probing how the genes interact, rather than investigating them in conceptual silos.
Coda
When people talk about the race between variants and vaccines, they aren’t being flip. For now, the vaccines stimulate a diverse enough antibody response to handle circulating viruses. But evolution never stops. If variants arise that slip into vaccinated bodies and take hold, then spread, will those vaccines weed out the older variants while creating niches for the new? That’s what is currently unsettling the experts. And me.
And so we must anticipate and stay ahead of evolution, which the vaccine manufacturers have already been doing for months. For if there’s one constant during these crazy times, it’s that SARS-CoV-2 continually surprises us. But for now, I’m relieved to ponder alternatives to the unthinkable idea that the virus was created to destroy us.
Ricki Lewis has a PhD in genetics and is a science writer and author of several human genetics books. She is an adjunct professor for the Alden March Bioethics Institute at Albany Medical College. Follow her at her website or Twitter @rickilewis